Metallocene CSTR
Reaction theory
The multiple metallocene CSTR reaction theory uses an algorithm based on the reaction scheme given by Read and Soares [1]. That paper presented some analytical and semi-analytical derivations of molecular weight distributions for the case of two metallocene catalysts. The present algorithm is a Monte Carlo algorithm for simulating the case multiple catalysts. Note that some catalysts behave non-ideally, giving broad molecular weight distributions. These will need to be modelled as a combination of several catalysts with different rate parameters.
Simulation parameters
On opening the theory, one is presented with (apparently) only four parameters, which are:
num_to_make:controls the number of molecules made in the simulation - more molecules mean better statistics, but take up more memory and take longer to simulate! It is a good idea, when trying to match data, to start with just a few molecules - say 1000 to 10000 - and then increase this number when you are satisfied the parameters are close to where you want them.
mon_mass:this is the mass, in a.m.u., of a monomer (usually set to 28).
Me:the entanglement molecular weight - needed for output to a BoB polymer configuration file, but has no effect on the display within the React module.
nbins:this is the number of bins - equally spaced in \(\log_{10}\text{MW}\) - used for analysis of the molecules. More bins means more resolution in the \(\log_{10}\text{MW}\) axis, but also more noise because there are fewer molecules per bin. Thus, the quality of the curves produced is a compromise between these two factors. Usually 50 bins is quite adequate for a reasonable number of molecules.
The remaining parameters are shown when you press the  button, which
opens a form looking like:
button, which
opens a form looking like:
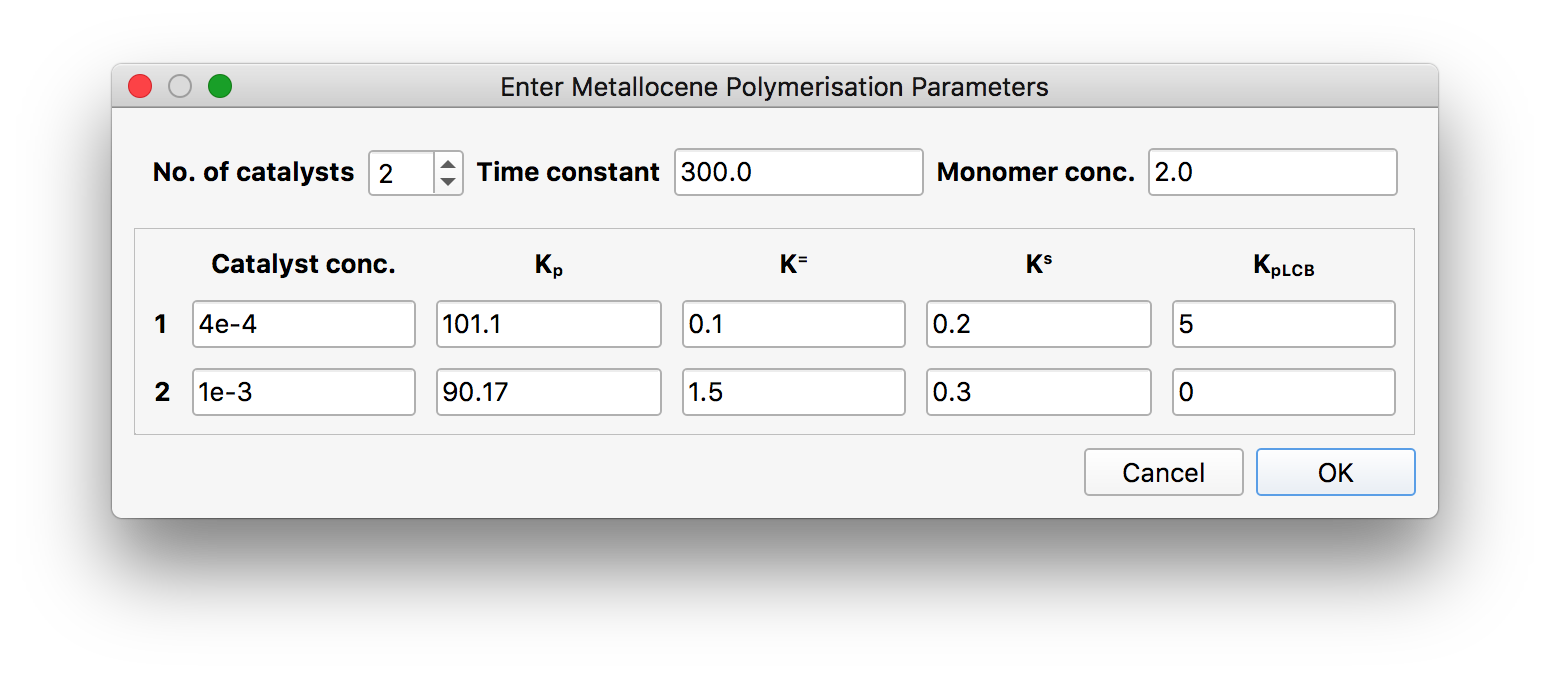
Here, one can set the total number of catalyst sites to be used in the calculation, the reactor time constant (the mean residence time in the CSTR), and the reactor monomer concentration. Then, for each catalyst site, there are five parameters to be fixed, as described in [1]:
The active catalyst site concentration (if catalyst deactivation is significant, one should account for this by reducing this parameter).
The polymerisation rate constant, \(K_\text{p}\).
The rate constant for chain transfer to macromonomer, \(K^=\). Some chain transfer reactions result in the creation of a macromonomer, which can then subsequently be incorporated into a growing chain, forming a long-chain-branch. This chain transfer rate constant describes a reaction of form
\[P\rightarrow D^{=}+C,\]where \(P\) is a macromonomer and \(C\) is a free catalyst site (or short growing chain). Thus, \(K^=\) is the total rate constant for all such processes, and the concentration of any chain transfer agents should be included within this rate constant.
The rate constant for chain transfer to dead chains, \(K^\text{s}\). Some chain transfer reactions result in the creation of a dead chain, which plays no further part in the reaction. Similar comments apply as for the constant \(K^=\)
The polymerisation rate constant for incorporation of macromonomers to form long-chain-branches, \(K_\text{p,LCB}\).
When a molecule is generated, it is straightforward to assess its molecular weight and number of branches.
Warning
The g-factor is calculated based on the assumption of ideal random walk statistics (as opposed to self-avoiding walk statistics). Ideal chain radius of gyration is fast to calculate: self-avoiding walk radius is not. It is often found that the g-factor calculated in these two ways is (somewhat surprisingly) similar, but it is still a possible source of error!
Polymer storage and saving to BoB
When a molecule is generated using the algorithm, the theory makes a decision as to whether to “save” the molecule (that is, to store a complete record of the molecule, including the connectivity of all the arms) or not (for an unsaved molecule, the theory still retains a record of the total molecular weight, number of branches and g-factor).
“Saved” molecules are retained for possible output into a BoB polymer configuration file. The decision as to whether to save a molecule, or not, is based upon whether there have been a given number of molecules of similar molecular weight already generated. The \(\log_{10}\text{MW}\) axis is split into a set of evenly-spaced bins, and each bin keeps track of how many polymers have been made with molecular weight in the range of that bin. Once the number made exceeds a given maximum, polymers of that molecular weight are no longer saved. However, the algorithm still keeps track of the number of polymers made in a given bin - when saving a polymer configuration file for BoB, the weights of the saved molecules are adjusted accordingly, to account for the unsaved molecules.
You can modify the parameters used for saving molecules by clicking on the
 button. This opens a form with four parameters:
button. This opens a form with four parameters:
the maximum molecular weight of the bins,
the minimum molecular weight of the bins (it is wise to make sure these span the MW range of the polymers you are making),
the number of bins,
the maximum number of polymers stored per bin.
In particular, increasing either of the last two parameters increases the number of polymers saved.
Clicking the  button opens a save dialogue box which allows you to save a polymer
configuration file containing the connectivity for the saved polymer. The
format of this file, and the use of it within a BoB calculation, is given in
the BoB documentation.
button opens a save dialogue box which allows you to save a polymer
configuration file containing the connectivity for the saved polymer. The
format of this file, and the use of it within a BoB calculation, is given in
the BoB documentation.
Memory issues
React stores polymer information (total molecular weight, number of branches and g-factor) in a polymer record, and connectivity information in arm records.
Note
There is a fixed amount of computer memory allocated for this. React is designed to cope smoothly with running out of memory - it should kindly ask you to allocate more memory! The dialog will propose you to increase the amount of computer memory allocated to React and give you an estimate the extra amount or RAM needed.
Tip
There are some things that contribute to using a lot of memory:
Some choices of reaction parameters lead to extremely large molecules, with lots of arms being generated on each molecule. For non-zero values of the parameter \(\beta\), gelation (i.e. infinite molecules) is possible. If a molecule looks like being particularly large, you will get a warning message!
If you make too many molecules, you will run out of memory.
If you save too many molecules, you will run out of arm records (adjust the parameters by clicking on the
button.If you have other theories open which are already using a lot of storage, there might not be enough memory left for your current calculation (close un-needed theories, or adjust their parameters so they don’t use as much storage).
You will get brief hints along these lines in the theory log if you run out of parameters.
References
Daniel J. Read and Joao B P Soares. Derivation of the Distributions of Long Chain Branching, Molecular Weight, Seniority, and Priority for Polyolefins Made with Two Metallocene Catalysts. Macromolecules, 36(26):10037–10051, 2003. doi:10.1021/ma030354l.